クロイツフェルト・ヤコブ病は、脳に迅速かつ漸進的に、そして深刻な影響を与えるまれな神経変性疾患です。
クロイツフェルト・ヤコブ病(CJD)は徐々に脳細胞を破壊し、脳に小さな穴ができます。 CJDの人々は、運動失調、体の動き、異常な歩行、発語、および認知症を制御することが困難である。
それは常に致命的であり、治療法はありません。
CJDは、米国(米国)を含め、世界中で毎年百万人に1人に影響を及ぼします。
原因は散発的、遺伝的、または取得される可能性があります。それは主に60歳以上の人々に影響を及ぼし、30歳未満の人ではまれです。
CJD患者は、症状が現れてから1年以上生存することはまずありません。
CJDとは何ですか?
![[CJD]](https://demedbook.com/images/1/what-is-creutzfeldt-jakob-disease-cjd.jpg)
CJDは伝播性海綿状脳症(TSE)であり、経時的に脳を破壊する。プリオンと呼ばれる病原体が原因です。プリオンはウイルスや細菌ではありません。
TSEの他のタイプには、Gerstmann-Strussler-Scheinker(GSS)症候群、致命的な家族性不眠症、およびヒトのクルーが含まれる。その他の例としては、羊や山羊のスクレイピー、ウシ海綿状脳症(BSE)、あるいはウシの「狂牛病」などがあります。
類似の脳症および消耗症候群が他の種に見られ、それらは実験動物において伝達可能であることが示されている。
疾病管理予防センター(CDC)は、古典的なCJDはBSEや他の亜種には関連していないことに注意しています。
症状
CJDは長い潜伏期間を有する。症状が現れるまでに最大40年かかることがあります。脳細胞が破壊されると症状が起こります。患者の状態は数週間で急速に悪化し、ほとんどの人は1年以内に死亡する。
CJDの顕著な症状は、認知症およびミオクローヌスへの急速な進行、または筋肉群の痙攣性の不随意運動である。
気分や行動の変化、性格の変化、記憶喪失、および判断の障害が一般的です。アルツハイマー型認知症やハンチントン病に似ているかもしれませんが、症状は数年ではなく数日から数週間で進化します。
病気が進行するにつれて、協調およびミオクローヌスの問題が悪化し、視力が損なわれて失明に至る。最終的に、患者はもはや動くことも話すこともできず、昏睡状態に入る。
脳組織の剖検は、CJDが他の認知症に見られないいくつかの独特の変化を含むことを明らかにした。
古典的なCJDに必ずしも関連していないCJDのいくつかの変種があり、病気の症状および経過は異なる可能性がある。
原因
CJDは、異常な種類のアミロイドタンパク質であるプリオンタンパク質が他のタンパク質に異常を引き起こすときに起こります。脳細胞上のプリオンの蓄積および異常は、最終的に脳の損傷および死につながる。
散発的、遺伝的、または取得される可能性があります。
142214
散発性CJD
症例の85%において、CJDは散発的である。明らかなリスク要因はない。
継承されたCJD
症例の5%〜10%が継承されます。それらは、プリオンタンパク質の形成を制御する遺伝子に変化が生じたときに起こる。 CJDの家族歴があるか、または卵子または精子細胞に突然変異が起こり、子孫がその病気を発症する危険性があります。
プリオンは遺伝情報を含まず、それらを再現するための遺伝子は必要ないが、体内の正常なプリオンタンパク質の遺伝子の変異は、プリオンを異常に働かせる原因となる。
プリオン遺伝子のいくつかの異なる突然変異が同定されている。各家族に見られる特定の突然変異は、その病気の出現頻度およびどの症状が最も顕著であるかに影響を及ぼす。
プリオンタンパク質遺伝子の突然変異を持つ全員がCJDを発症するわけではありません。
CJDを買収
どんな種類のCJDもある人から他の人に渡すことができるという証拠はありませんが、いくつかの手順はCJDの伝達と関連しています。
これらには、
- 角膜移植
- 電極インプラント
- 硬膜外移植片、または髄膜移植片であり、これは脳の外皮の移植片である
- ヒト成長ホルモンの使用
約1%の症例が、罹患した脳または神経系組織への既知のまたは高度に疑われる曝露によって伝達される。
ウシ海綿状脳症
1990年代にCJDの1つのタイプが牛で発生するBSEへの曝露に関連していました。
![[プリオン理論]](https://demedbook.com/images/1/what-is-creutzfeldt-jakob-disease-cjd_2.jpg)
伝染は食物消費に関連していると考えられていた。この変異体は若年患者に影響を与える傾向があり、さらに長期間持続した。
BSEは、ウシ、ヒト、およびネコを含む多くの種に影響を及ぼします。
一部の科学者は、異常な「遅いウイルス」や他の生物がCJDを引き起こすと信じていますが、まだその病気の人に特定のウイルスや生物を隔離していません。
CJDの原因となる薬剤には、ウイルスやバクテリアでは通常ないいくつかの特徴があります。
これには、長いインキュベーション期間、殺すことが困難であるという事実、核酸、DNAまたはRNAの形で遺伝情報を含まないように見えることが含まれる。
科学者たちは、CJDおよび他のTSEが、生きている生物によってではなく、プリオンによって引き起こされると信じている。プリオンは生きていませんが、脳内で拡大する異常な構造のタンパク質です。
この膨張は脳組織を損傷し、CJDの特徴的な症状を引き起こす。
診断
CJDの診断を確認する試験はありません。脳生検のみがこれを行うことができ、生存中は患者にとって危険です。
テストは、最も可能性の高い原因を見つけるのに役立ちます。
身体検査で筋痙攣を探し、患者の反射をチェックする。これらは通常より反応性が高いかもしれません。病気が脳にどこに影響を及ぼすかに応じて、筋肉は過度に調子が悪くなったり枯れたりすることがあります。
視力検査または眼球検査は、患者が以前に気付いていなかった部分的な失明を検出することができる。
脳波(EEG)は、異常な電気インパルスを明らかにすることができる。
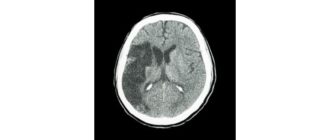
CTスキャンまたはMRIは、症状の原因として脳卒中を除外することができる。
腰椎穿刺または脊髄穿刺は、脊髄液を検査して痴呆の他の原因を排除することができる。中枢神経系(CNS)に感染があるか圧迫されているかを示すことができます。
タンパク質14-3-3が液体中に見出され、その人が典型的な症状を示している場合、その人にCJDがある可能性が高い。
死後の脳生検では、脳組織がスポンジ状であり、神経細胞の塊が破壊されたところに小さな穴が見えることが示されている。
処理
CJDの治療法はありませんし、薬を投与することでコントロールすることも、病気の進行を遅らせることもできません。
治療は、症状を緩和し、患者をできるだけ快適にすることを目的とする。
オピエート薬は痛みを和らげるのに役立ちます。クロナゼパムとバルプロ酸ナトリウムは、筋痙攣などの不随意運動を緩和するのに役立ちます。
後の段階では、患者は頻繁に移動して褥瘡を予防する必要があります。尿を排出するためにカテーテルを使用することができ、授乳は静脈内の液体によって行われる。
防止
予防措置には、病気の原因となる可能性のある生物を殺すためのすべての医療機器の滅菌、CJDの診断または可能性のある人の角膜献血を受け入れないことが含まれます。
ほとんどの国で、他の形態のTSEがヒトに伝播する可能性を避けるため、感染した牛の管理と飼料に関する制限について厳しいガイドラインが作成されています。
CJDの診断を受けた個人に曝されている人は、いくつかのガイドラインに従うように勧められます。
これらには、
- 開いた創傷、切れ目、および擦り傷を皮膚に被覆する
- 患者の組織、血液、または液体を取り扱う際に手袋を着用する
- 患者との接触のために使い捨てのガウンまたは衣服を着用する
- 汚染された液体が飛散する危険性がある場合は、顔面シールド、目の保護具、またはマスクを使用する
- 患者の上または近くで使用された殺菌装置
脳腫瘍のプリオンの役割、脳にどのように影響を与えるのか、そして効果的な治療法を見つけるための研究が進められています。