臨床試験は、医療戦略、治療、またはデバイスが人間の使用または消費に対して安全であるかどうかを判断することを目的とした調査研究です。
これらの研究はまた、特定の条件や人々の集団に対する医療的アプローチがどれほど効果的であるかを評価するかもしれない。
全体として、彼らは医療知識を増やし、信頼できるデータを提供して、医療の意思決定とガイドラインを支援します。
参加者の安全を確実にするために、小規模なグループから試験を開始し、新しい方法が害を及ぼすかどうか、または不満足な副作用があるかどうかを調べます。これは、実験室や動物で成功した技術が人にとって安全で効果的でない可能性があるためです。
臨床試験に関する迅速な事実
- 臨床試験は、医療戦略、治療、またはデバイスが人間が使用または消費するために安全かつ効果的であるかどうかを調べることを目指しています。
- 試験は4つのフェーズで構成され、治療、予防、診断、スクリーニング、支援ケア、保健サービス研究、基礎科学に焦点を当てることができます。
- 研究チームには、医師、看護師、ソーシャルワーカー、医療従事者、科学者、データ管理者、臨床試験コーディネーターが含まれる可能性が高い。
- 参加には、リスクと利益の両方が含まれる可能性があります。参加者は、トライアルに参加する前に「インフォームドコンセント」文書を読み、署名する必要があります。
- リスクは管理され監視されていますが、医学研究の本質はいくつかのリスクが避けられないことを意味します。
臨床試験は何ですか?
臨床試験の主な目的は研究です。治験は、疾患または状態の治療、診断および予防に関連する医学的知識を追加するように設計されている。

研究は以下を目指す厳しい科学的基準とガイドラインに従います:
- 参加者を保護する
- 信頼性の高い正確な結果を提供する
人間に対する臨床試験は、長く、体系的で、徹底した研究プロセスの最終段階で行われます。
このプロセスは、新しいコンセプトが開発され、テストされるラボで開始されることがよくあります。
動物を試験することで、科学者はそのアプローチが生体にどのように影響するかを見ることができます。
最後に、人間のテストは、小規模で大きなグループで実施されます。
トライアルは次の目的で実施することができます。
- 疾患、症候群、または状態(例えば、薬物、医療機器、または手術または療法へのアプローチ)についての1つまたは複数の治療介入を評価する。
- 例えば、医薬品、ワクチン、生活習慣の変化などによって疾患や状態を予防する方法を評価する
- 特定の疾患または状態を特定または診断する可能性のある1つ以上の診断介入を評価する
- その状態の認知のための識別方法またはその状態の危険因子を調べる
- 慢性疾患の人々の快適さと生活の質を改善するための支援的ケアの手順を探る
臨床試験の結果は、新しい医療戦略、治療またはデバイスがあるかどうかを特定することができます。
- 患者の予後に正の効果を有する
- 予期しない害を引き起こす
- 肯定的な利益を持たないか、または悪影響を有する
臨床試験は、治療の費用対効果、診断テストの臨床価値、治療がQOLを向上させる方法に関する貴重な情報を提供します。
臨床試験の種類
すべての臨床試験には主な目的があります。これらは、次のカテゴリに分類できます。
- 治療:新しい治療法、新薬の組み合わせ、または手術または治療への新しいアプローチのテスト
- 予防:医薬品、ビタミン、ワクチン、ミネラル、生活習慣の変化などを通じた疾病の予防または再発を改善する方法の検討
- 診断:病気や症状を診断するための改善された検査技術と手順の発見
- スクリーニング:特定の病気や健康状態を特定するための最良の方法のテスト
- 支持療法:慢性疾患患者の快適性と生活の質を向上させるための手続きの調査
- 保健サービス研究:ヘルスケアの提供、プロセス、管理、組織、または資金調達の評価
- 基礎科学:介入のあり方の検討
臨床試験はなぜ重要ですか?
臨床試験は医療の改善と進歩を助ける。これらの研究は、患者のケアを改善するために使用できる事実上の証拠を提供する。
臨床研究は、医師が次のような要素を認識していない場合にのみ実施されます。
- 新しいアプローチが人間で効果的に働き、安全であるかどうか
- 特定の病気やグループに最も効果的な治療法や戦略
臨床試験はどのように機能しますか?
臨床試験の設定、実行、フォローアップにはさまざまな要素が関わっています。
臨床試験プロトコール

試験は包括的な計画、すなわちプロトコルに従う。プロトコルとは臨床試験の記述です。
それには、研究の目的、設計と方法、関連する科学的背景、統計情報が含まれます。
含める主な情報は次のとおりです。
- 参加者数
- 誰が参加する資格があるか
- どのようなテストが行われるか、どのくらいの頻度で
- 収集するデータの種類
- 研究の長さ
- 治療計画に関する詳細情報
偏見を避ける
研究者は偏見を避けるための対策を講じる必要があります。
バイアスとは、プロトコールに関係しないが、試験の結果に影響を与える可能性のあるヒトの選択または他の因子を指す。
バイアスを回避するのに役立つ手順は、比較グループ、ランダム化、およびマスキングです。
比較グループ
ほとんどの臨床試験では、比較グループを使用して医療戦略と治療法を比較しています。あるグループが他のグループよりも優れた結果を示すかどうかを示す結果が表示されます。
これは通常、次の2つの方法のいずれかで実行されます。
- 1つのグループは、ある状態のための既存の治療を受け取り、第2のグループは、新しい治療を受ける。研究者はどのグループが良い結果を持っているかを比較します。
- 1つのグループは新しい治療を受け、第2のグループは試験製品のような不活性な製品であるプラセボを受けます。
ランダム化
比較グループとの臨床試験では、しばしばランダム化を使用します。参加者は、選択肢ではなく偶然、比較グループに割り当てられます。これは、試行中に見られる差異は、使用された戦略によるものであり、参加者間の既存の差のためではないことを意味する。
マスキングまたは目隠し
マスキングや目立たないことは、参加者や研究者のいずれに治療を受けようとしているのかを通知しないことによって偏見を避けるのに役立ちます。
シングルブラインド:これは、参加者または研究者がどちらのグループであるかを認識していない場合です。
二重盲目:これは、参加者と研究者の両方が気づかないときです。
混乱要因
コンファウンダーは、2つ以上の特性間の真の関係を歪める可能性があります。
例えば、たばこライターを携行する人々は、軽い方の持ち運びにより肺がんを引き起こすため、肺がんを発症する可能性がより高いと結論づけることができます。この例では、喫煙は交絡相手である。
シガーライターを持っている人は喫煙者である可能性が高く、喫煙者は肺がんを発症する可能性がより高いですが、他の目的のためにライターを持ち運ぶ人もいます。
これを考慮しないと、偽の結論につながる可能性があります。
研究チームには誰がいますか?
通常は医師である主治医が各臨床試験をリードする。
研究チームには、
- 医師
- 看護師
- ソーシャルワーカー
- ヘルスケアのプロ
- 科学者
- データマネージャー
- 臨床試験コーディネーター
臨床試験はどこで実施されていますか?
場所は、研究の種類と誰がそれを整理しているかによって異なります。
いくつかの一般的な場所が含まれます:
- 病院
- 大学
- 医療センター
- 医師のオフィス
- コミュニティクリニック
- 連邦政府および産業基金の研究サイト
どのくらいの試練が続くのですか?
これは、他の要因の中で、何が調査されているかによって異なります。いくつかの試験は最終日であり、他の試験は数年続く。
試用に参加する前に、参加者はどれくらいの期間続くことが予想されるかを聞かれます。
設計と組織
さまざまな種類の学習と、それらを整理するさまざまな方法があります。ここにいくつかの研究タイプがあります。
観察研究
コホート研究および症例対照研究は、観察研究の例である。
コホート研究

コホート研究は、研究集団またはコホートを選択する観察研究である。
どの科目にどちらかの科目があることを確認するための情報が収集されます。
- 問題の疾患の発症に関連すると考えられる血液型などの特定の特性
- 例えばタバコの喫煙のような疾患に関連する可能性のある因子への暴露
彼らは喫煙するので個人を選ぶことができます。その後、彼らは他の人々と比較して、彼らが病気を発症する可能性があることを時間通りに追跡することができます。
このタイプの研究は、肺がんへの喫煙の影響など、実験的に制御できない疑わしい危険因子の影響を研究するために使用されます。
コホート研究の主な利点は次のとおりです。
- 暴露は疾患発症前に測定されるため、疾患発症に関して偏見がない可能性がある。
- まれな曝露は、研究コホートの適切な選択によって調査することができる。
- 任意の1つの暴露について、複数のアウトカムまたは疾患を研究することができます。
- 病気発生率は、暴露群と非暴露群の両方で算出することができる。
コホート研究の主な欠点は次のとおりです。
- それらは、特に前向きに実施される場合には、高価で時間がかかる傾向があり、これは前進することを意味する。
- 曝露状況および診断基準の両方の経時変化は、曝露および病状に応じて個体の分類に影響を及ぼす可能性がある。
- 被験者の曝露状況が分かっているため、結論に情報バイアスが存在する可能性がある。
- フォローアップへの損失は選択バイアスをもたらす可能性がある。
症例対照研究
症例対照研究は、特定の病状のリスク要因を区別することができる。
研究者は、状態のある人とそれがない人を比較する。時間を遡って作業することで、2つのグループの違いを特定します。
症例対照研究は、結果から始まって曝露を調査するために追跡するため、後ろ向きに見て回顧的です。
症例対照研究の主な利点は次のとおりです。
- 発見はすぐに得ることができます。
- この研究は最低限の資金調達またはスポンサーシップで行うことができます。
- 彼らは、誘導期間が長い希少疾患や疾患の調査に効果的です。
- さまざまな可能性のあるリスク要因を検討することができます。
- 複数の曝露を研究することができる。
- 彼らはほとんどの研究対象を必要としません。
症例管理研究の主な欠点は次のとおりです。
- インシデントデータを生成することはできません。
- 彼らは偏見の対象です。
- 記録保持が不十分または信頼できない場合、過去の被ばくの正確で偏りのない測定値を得ることは困難であり得る。これは情報バイアスと呼ばれます。
- コントロールの選択は問題になる可能性があります。これは選択バイアスを導入する可能性がある。
- 曝露と疾患との間の年代順は同定するのが難しいかもしれない。
- 彼らは、曝露が大部分の症例の原因とならない限り、まれな曝露を調べるのには適していません。
ネストされた症例対照研究
ネストされた症例対照研究では、症例群および対照群は同じ研究集団またはコホートから来る。
コホートがフォローされているので、発生したケースがケースコントロールスタディの「ケース」になります。コホートの罹患していない参加者は、「コントロール」となる。
ネストされた症例対照研究は、コホート研究と比較して、コストが低く、時間の節約が少ない。
この疾患の発生率および有病率は、時々ネストされた症例対照コホート研究から予測されることがある。これは、単純な症例対照研究では不可能である。なぜなら、曝露された個体の総数および追跡期間は、通常知られていないからである。
ネストされた症例対照研究の主な利点は次のとおりです。
- 効率性:コホートの参加者のすべてが診断検査を必要とするわけではない。
- 柔軟性:彼らは、コホートが計画されたときには予想されなかった仮説のテストを可能にする。
- 選択バイアスの軽減:症例と対照は同じ集団からサンプリングされる。
- 情報バイアスの軽減:治験責任医師が症例の状態を知ることなく、リスクファクターの暴露を評価することができる。
主な欠点は、サンプルサイズが小さいため、結果の権限が低くなることです。
生態学的研究
生態学的研究は、人口または地域社会の暴露と結果との関係を調べる。
生態学的研究の一般的なカテゴリーには、
- 地理的比較
- 時系列分析
- 移行の研究
生態学的研究の主な利点は次のとおりです。
- 定期的に収集された健康データを利用できるので、安価です。
- 他の研究よりも時間がかかりません。
- それらは単純ではなく、理解するのが簡単です。
- 食事や大気汚染、温度などのグループや地域で測定されたばく露の影響を調べることができます。
生態学的研究の主な欠点は次のとおりです。
- 生態学的な誤りとして知られている控除の誤りが生じる可能性があります。研究者がグループデータの分析のみに基づいて個人に関する結論を導くときに起こります。
- 結果関係への暴露は検出するのが難しい。
- 交絡要因に関する情報が不足しています。
- 曝露の測定方法の分野間には体系的な違いがあるかもしれません。
実験的研究
観察研究とは別に、治療研究を含む実験的研究もある。
ランダム化比較試験

ランダム化比較試験(RCT)は、個体を無作為に割り付けて、特定の介入を受けるか受けないかのどちらかを割り当てる。
2つの異なる治療法の1つ、または治療法とプラセボが使用されます。
これは、どの治療法が最も効果的であるかを特定する最も効果的な研究タイプです。外部変数の影響を低減します。
RCTの主な利点は次のとおりです。
- 研究者の側には意識的または潜在的な偏見はありません。これは本質的に外部の妥当性を保証します。
- サンプルグループが十分に大きければ、年齢、性別、体重、活動レベルなどの混乱する変数を取り消すことができます。
RCTの主な欠点は次のとおりです。
- 彼らは時間がかかります。
- 彼らは高価なことができます。
- 彼らには大きなサンプルグループが必要です。
- まれな出来事は勉強が難しいことがあります。
- 偽陽性および偽陰性の両方の統計的エラーが可能である。
適応型臨床試験
適応設計手法は、収集されたデータに基づいています。それは柔軟で効率的です。治験および進行中の臨床試験の統計的手順を変更することができる。
準実験
準実験的または無作為化された研究には、無作為化されていない幅広い介入研究が含まれています。このタイプの試験は、RCTが論理的に実行可能でないか倫理的でない場合に頻繁に使用されます。
証拠の階層
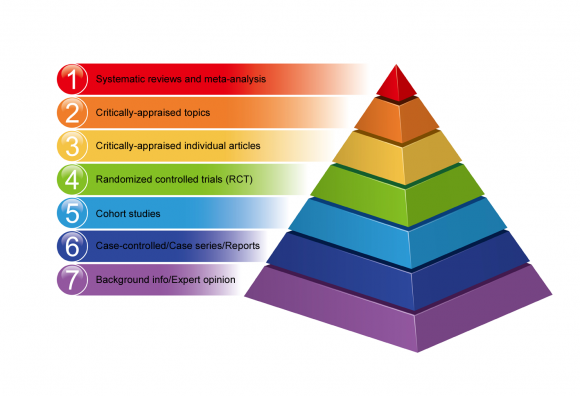
証拠の階層構造により、調査結果の妥当性に応じてさまざまな調査方法のランク付けが可能になります。
すべての研究デザインが、エラーのリスクと結果の偏りの点で同等であるわけではありません。いくつかの研究方法は他の研究方法より優れた証拠を提供する。
下にある証拠の質の低さから上の高品質の証拠まで、ピラミッドの形で証拠に基づく医療の階層の例を以下に示します。
臨床試験の段階
医学研究の研究は、段階と呼ばれるさまざまな段階に分かれています。薬物検査の場合、これらはFDAによって定義されています。
早期相試験は、薬物の安全性およびそれが引き起こす可能性のある副作用を調査する。後の試験では、新しい治療法が既存の治療法よりも優れているかどうかが検査されます。
第0相試験:薬力学および薬物動態
フェーズ0は、早い段階で新薬の臨床情報を提供するのに役立つ探索段階です。
このフェーズ:
- フェーズ1の早期に実施される
- 非常に限られた人間の暴露を伴う
- スクリーニングおよび微量投与研究に限定されている治療または診断の意図はない
フェーズ1試験:安全性のスクリーニング
フェーズ0の後、人間にはさらに4つの試験段階があります。これらはしばしば重複します。段階1〜3は、ライセンスが付与される前に行われます。
フェーズ1のガイドラインは次のとおりです。
- 健康なボランティア20人と80人の間
- 薬物の最も頻繁な副作用の確認
- 薬物がどのように代謝されて排泄されるかを見出す
フェーズ2の試験:有効性の確立
第1相試験が許容できない毒性レベルを示さない場合、第2相試験を開始することができる。
これには、
- 36人から300人の参加者
- 特定の疾患または状態の人々に薬剤が働くかどうかに関する予備的なデータを収集する
- 同様の状況下で、異なる薬物またはプラセボを服用している人々と薬物を摂取している被験者とを比較するための対照試験
- 継続的な安全評価
- 短期間の副作用に関する研究
フェーズ3の試験:安全性と有効性の最終確認
フェーズ2で薬の有効性が確認されたら、FDAとスポンサーはフェーズ3で大規模な研究を行う方法について話し合う。
これには以下が含まれます:
- 300〜3000人の参加者
- 安全性と有効性に関するさらなる情報を収集する
- 異なる集団の研究
- 最高の処方量を決定するために様々な投薬量を調べる
- 有効性を判断するために他の薬物と組み合わせて薬物を使用する
この段階の後、新薬に関する完全な情報が保健当局に提出されます。
審議会
FDAがマーケティングのために製品を承認すると、市販後の要件とコミットメントスタディが実施されます。
FDAは、これらの試験を使用して、製品に関するさらなる安全性、有効性または最適な使用情報を収集します。
新薬申請

ドラッグスポンサーは、新薬申請(NDA)を完了し、米国でのマーケティングのために新薬の承認を検討するようFDAに要請する。
NDAに含まれるもの:
- すべての動物および人間のデータ
- データの分析
- 体内の薬物行動に関する情報
- 製造詳細
FDAは、それを審査するかどうかを決定するのに60日間を要する。
彼らがNDAを提出することを決定した場合、FDA審査チームは、スポンサーの薬物の安全性および有効性に関する研究を評価するように任命される。
次の手順を実行する必要があります。
医薬品表示:FDAは、医薬品の専門的な表示を確認し、適切な情報が消費者および医療従事者と共有されることを確認する。
施設検査:FDAは、医薬品が製造される施設を検査する。
医薬品承認:FDA審査員は、申請を承認するか、または応答書を発行する。
フェーズ4の試験:販売中の調査
第4相臨床試験は、薬剤の販売承認後に行われます。彼らは以下を含むように設計されています:
- 1,000人以上の患者
- より大きなグループおよび患者の亜集団における新薬の安全性および有効性の評価における包括的な経験
- 他の利用可能な治療との比較および組み合わせ
- 薬物の長期的な副作用の評価
- あまり一般的でない有害事象の検出
- 他の伝統的療法および新しい療法と比較して薬物治療の費用効果
安全性報告書
FDAが薬物を承認した後、販売後の段階が始まります。スポンサー、通常メーカーは、FDAに対して定期的な安全性の更新を提出します。
誰が臨床試験を後援しますか?
臨床試験や研究には数億ドルのコストがかかることがあります。試行の資金を提供するグループには、
- 製薬、バイオテクノロジー、医療機器企業
- 学術医療センター
- 自主的なグループと財団
- 国立衛生研究所
- 政府部門
- 医師および医療提供者
- 個人
誰が参加できますか?
このプロトコルは、誰が試験に参加する資格があるのかを定義します。
可能性のある包含基準は、
- 特定の病気または状態を有する
- 健康状態ではなく「健康」である
除外基準は、一部の人々が試験に参加することを排除する要因です。
例には、年齢、性別、疾患の特定の種類または段階、以前の治療歴および他の病状が含まれる。
考えられる利点とリスク
臨床試験に参加することで、参加者にとって利益とリスクの両方を得ることができます。
臨床試験のメリットとしては、以下が挙げられます。
- 参加者は新しい治療法にアクセスできます。
- 治療が成功すると、参加者は最初に利益を得ることになります。
- 新しい治療を受けているグループに参加していない参加者は、特定の状態の標準治療を受けることがあります。これは新しい治療法と同じかそれ以上です。
- 健康は密接に監視され、医療機関のチームによってサポートされます。
- 臨床試験から集められた情報は、科学的知識を増やし、他者を助けるかもしれないし、最終的に医療を改善する。
考えられるリスクには、
- 特定の状態に対する標準的なケアは、時には、研究されている新しい戦略または治療法よりも優れている場合があります。
- 新しいアプローチや治療は、一部の参加者にとってはうまくいくかもしれませんが、必ずしも他の参加者にとってはうまくいかないかも
- 予期せぬ予期せぬ予期せぬ副作用、特に第1相試験および第2相試験、および遺伝子療法または新しい生物学的治療などのアプローチがあるかもしれません。
- 健康保険および保健医療提供者は、常に臨床試験に参加している患者のケアおよび費用をカバーしているわけではありません。
同意を与えることはどういう意味ですか?

インフォームドコンセント文書には、臨床試験に参加する際のリスクと潜在的な利点が説明されています。
ドキュメントに表示されなければならない要素には、
- 研究の目的
- 予見可能な不快感のリスク
- 可能な利点
参加者は同意書を徹底的に読んで、トライアルに参加される前に登録と署名をしたいかどうかを判断する必要があります。
臨床試験は安全ですか?
FDAは、試験に参加することを検討している誰でも、リスクに関する情報を含め、情報に基づいた選択を行うために必要なすべての信頼できる情報にアクセスできるようにしています。
参加者へのリスクは管理され、監視されますが、医学研究の性質上、いくつかのリスクは避けられません。
参加者はどのように保護されますか?

参加者の安全が最優先事項です。すべての試行において、科学的な監督と患者の権利が彼らの保護に貢献します。
グッド・プラクティス・プラクティス(GCP)は、治験において倫理的かつ適切な処置が確実に行われるようにすることを目的としています。
GCPの遵守は、参加者の安全と権利が保護されているということを一般市民に提供します。
これは、
- 参加者の権利、安全、福祉を守るため
- 収集されたデータが信頼性が高く、完全性があり、適切な品質であることを保証する
- 臨床研究の実施に関するガイドラインと基準を提供する
GCPの基礎は、1947年に最初に計画された。主なポイントは、どの試験でも、研究者は以下のことを保証しなければならないということであった:
- 自発的参加
- インフォームドコンセント
- リスクの最小化
時間の経過と共に、脆弱な集団の保護を強化し、研究を実施する団体に指導を提供するまで、追加が行われました。
患者の権利
患者の権利を保護する方法は次のとおりです。
インフォームドコンセントは、臨床試験参加者に試験のすべての事実を提供するプロセスです。参加者が参加することに同意する前に、そして試行中に発生します。インフォームドコンセントには、治療や受診可能性のある可能性のある利点やリスクに関する詳細が含まれています。
その他の権利:インフォームドコンセント文書は契約ではありません。参加者は、試行が完了したかどうかに関係なく、いつでも研究から離脱することができます。
子供の権利と保護:18歳以下の子供は、親または法定の保護者が法的に同意する必要があります。試行が最小限以上のリスクを伴う場合、両親は許可を与えなければなりません。 7歳以上の子供は、臨床試験に参加することに同意する必要があります。
臨床試験を見つけるにはどうすればよいですか?
現在の臨床試験に関する情報は、ここにあります。